VMD Selection Tool: How to Select Atoms and Residues in VMD
While observing and analyzing the overall structures of biomolecules using VMD, there comes a point when you want to focus your actions on specific subsets of atoms within the system.
For instance, you might wish to highlight a particular residue or emphasize the binding site of a protein using different representations or colors. To achieve this precision, VMD provides a powerful selection tool that enables you to interact with specific segments of the molecule by selecting them. Once selected, these subsets can be utilized to perform targeted actions, allowing for a more nuanced analysis of your molecular system.
In this article, you will learn how to use the selection tool in VMD to select atoms and residues in various ways. By the end, you will be ready to write your selections and apply these tips to your own system.
Access the Graphical Representations Menu
The initial step towards creating a new selection is to open the graphical representations menu. Here’s how to do it:
This is where you’ll find an overview of all available selections and the currently active ones.
Create Rep or Delete Rep to add or remove selections.
For a clearer understanding, take a look at the image below. In the red box, you’ll spot two current selections, each defined by its representation, color, and the selection criteria applied to the atoms. Meanwhile, the blue box points out the area where the current selection is showcased.
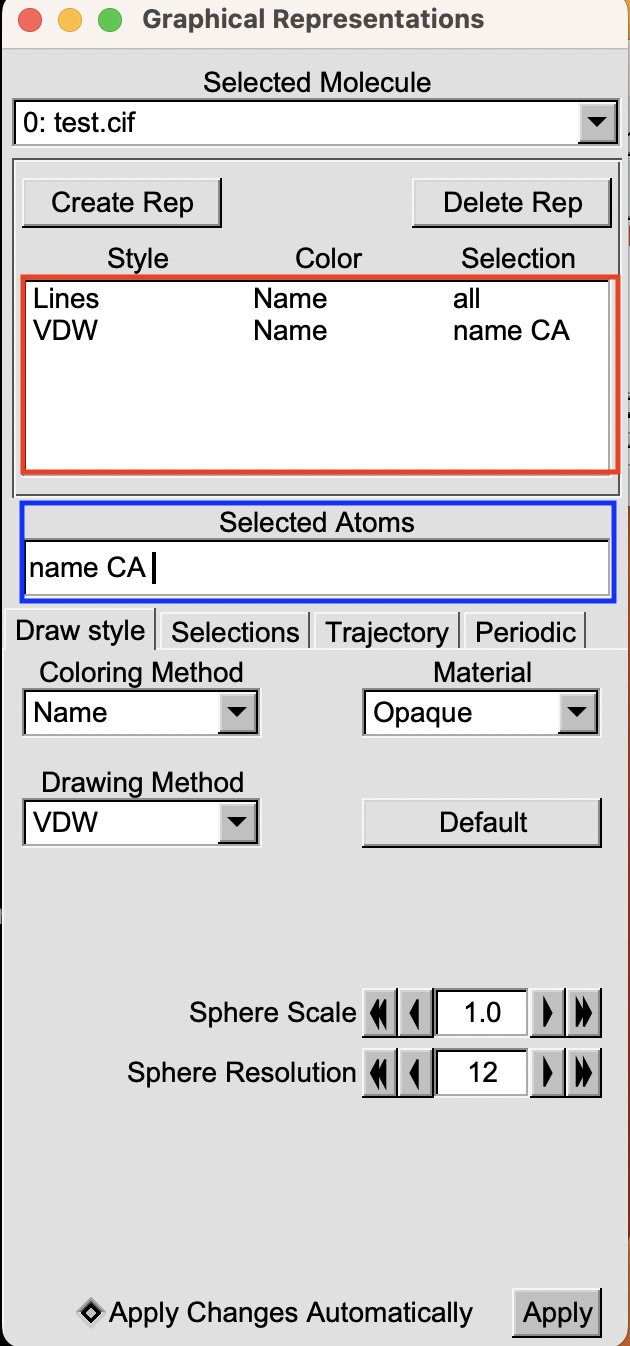
If you want to know how to change the appearance of a selection here is an article where I explain how to do this. Here we will just focus on how to select the atoms you are interested in (e.g. the. blue box).
Selecting Atoms in VMD
Something that you may find useful is to select certain atoms in particular that you want to use to modify their representation or color.
To select atoms in VMD you have many different options. Here I will go through the ones I generally use more often.
Select by Index
One thing you can do is to select by index. For instance, if you want to select atom number 100, you can type:
|
|
If you want to select a range of atoms, let’s say from 1 to 10 you can type:
|
|
Select by Name
Another option would be to select by name. For instance, if you want to select the alpha carbons in a protein, and you know that their atom name is CA, then you can execute:
|
|
This will select all atoms named CA e.g., all alpha carbons.
Select by Element
Lastly, you can also select atoms based on their elemental composition. This method enables you to focus on atoms of a particular element, such as carbon (C), hydrogen (H), oxygen (O), Nitrogen (N), and so on.
|
|
Selecting Residues in VMD
Another common option is to select residues in your protein. Also here you have different options, and I will go through some of them:
Selecting by Residue Number
If you know the specific residue number you wish to select, simply enter the command resid followed by the corresponding number. For example, to select residue 100, use the command:
|
|
Also in this case, if you want to select a range of residues, let’s say from 1 to 10 you can type:
|
|
Selecting by Residue Name
Another convenient method for selecting residues in VMD is based on their names.
If you aim to select all atoms within residues with a specific name, such as alanine (ALA) residues, you can use the command:
|
|
Similarly, this method applies to non-protein components as well. For example, if you have a ligand named LIG and you wish to select it, simply type:
|
|
General Selections in VMD
VMD offers a range of native keywords that simplify the selection process for various molecular components.
For instance, if you’re looking to select all protein atoms, you can effortlessly use the keyword:
|
|
This will select all atoms belonging to a protein.
Similarly, to select water molecules, you can employ the straightforward command:
|
|
You can also find more specific selections, such as isolating the backbone of residues (backbone), their sidechain (sidechain), or lipid atoms (lipid).
How to select atoms within a certain cutoff VMD
Another interesting feature of VMD is the possibility to select atoms within a certain distance from another one.
Maybe sometimes you would like to show all the residues within 5 Å of your ligand (LIG). In this case, you can say:
|
|
Similarly, if you’re interested in selecting aromatic residues within 5 angstroms of a specific residue number 100, you can employ:
|
|
Combine selection rules in VMD
Probably one of the most important things is the possibility of combining selection rules using Boolean operators. This functionality expands the scope of possible selections, enabling users to create more complex criteria in a single command.
For instance, suppose you want to select atoms with the name CA within the range of residues from 1 to 100. You can achieve this by combining selection rules using the and and to operators:
|
|
Similarly, to select every residue in the range of 10-20 or 30-40 in your protein, you can use the or operator to associate the keywords and select both residue ranges:
|
|
Finally, you can use the not logical operator to reverse the expression that immediately follows. For instance, if you want to select everything except alpha carbons, you can use:
|
|
How to use a VMD Selection
Using a VMD selection can be useful for many reasons.
The most obvious advantage is the ability to customize its appearance once you’ve created a new selection.
However, the usefulness of VMD selections extends beyond appearance customization. For example, you’ll encounter this selection tool in various functions, such as the tool used to perform RMSD analysis Here, you can specify different parts of your systems to compute RMSD selectively.
Additionally, VMD selections play a crucial role in tools like the one for hydrogen bond analysis. Here, specifying two selections enables the analysis of hydrogen bonds between specific segments.
These examples only scratch the surface of the myriad applications of VMD selections.