GROMACS Tutorial: Molecular Dynamics simulation of a protein in water environment
Background for molecular simulation
In this tutorial, we are going to see from a practical point of view how to simulate a protein in a water environment using GROMACS.
Most of the commands and functions needed to carry out a successful Molecular Dynamics (MD) simulation were already discussed in other articles.
I strongly suggest you read the article where I introduced GROMACS article before going through this tutorial so that you can have:
- A general idea of how GROMACS works.
- A proper understanding of the commands involved in a basic GROMACS simulation
Throughout the course of the tutorial, I will still redirect you to a more detailed description of the commands when needed. Feel free to click on those links to have a more clear explanation of each step.
I am going to assume that you have a general idea of the different concepts involved in a Molecular Dynamics (MD) simulation. I link you to a category of articles dedicated to the subject where you can find some useful concepts that you should be familiar with for a proper understanding of this tutorial.
A problematic aspect of GROMACS is that sometimes the simulation will run regardless of whether you have a clear idea about what you are doing or not. When you will start running your own simulations you will discover that it will be fundamental to know what is going on behind the scenes so that you can properly set your own parameters, and debug eventual errors.
I will also take for granted that you are more or less familiar with the Linux command line.
Having clarified that, we are ready for the tutorial.
System preparation
An MD simulation requires different steps. I will not go into the detail of each step, you can find more information about the different steps required to carry out an MD simulation here.
In the first step of the simulation, we need to select the protein and proceed to prepare the system for the experiment.
Protein Selection and initial setup
For this tutorial, we are going to consider a small structure that does not require extensive simulation times. The chicken villin subdomain will do just fine (PDB ID: 1YRF).
You can download the structure here, and then visualize it with your favorite visualization software. You can check this PyMOL introduction if you want more detail on how to visualize a molecular structure

First of all, we need to delete the water and the ligands from the pdb file. You can do it with your favorite text editor but the most common way to do it is to use the grep command like this:
|
|
Now we need to create a topology for our protein. To this aim, we are going to use the pdb2gmx command as follows:
|
|
gmx_mpi instead of the standard gmx.
GROMACS will ask us to select a Force Field (FF).
|
|
For this tutorial, we are going to Select “6” (AMBER99SB-ILDN)
We will generate three files from this run:
- A structure file (
1yrf.gro) - A topology file (
topol.top) - Position restraints (
posre.itp)
You can find more info on these file formats here
Define a box
At this stage, we need to create a virtual simulation box for our protein. You can do it with the editconf module.
|
|
We will need the protein to be at least 1.0 nm from the edge of the box to make sure that we don’t have problems when implementing Periodic Boundary Conditions (PBC).
You can load the resulting structure (1yrf_box.gro) with Pymol and use the command show cell to show the box containing the protein.

Solvate the system
Now we need to add solvent to the system through the solvate module. As already stated, we are going to use water as a solvent.
Type the following command:
|
|
You will generate a new gro file named 1yrf_solv.gro. If you open the file with Pymol you should see something that looks like Figure.

Neutralize
The last step of our system preparation phase consists of neutralizing the system by adding ions. This is accomplished via the genion module.
To use this module we first need a file containing the tpr file (ions.tpr) that is going to be used as input.
To generate it we need to use the grompp module. To use the grompp module we need an mdp parameter file (ions.mdp).
This may seem a little bit confusing if you are not familiar with GROMACS. This diagram may help to clarify the situation.
We need an ions.mdp file to use the grompp module. You can download it from here.
You can assemble the ions.tpr file with this command:
|
|
Now we can use this file as input for the genion module.
|
|
The genion module neutralizes the system by replacing some molecules in our system with ions.
GROMACS will ask us to select the molecules we want to replace with ions. We want to select the solvent (water molecules in our case) so that they will make place for the ions..
|
|
Select the solvent “13 SOL”.
The resulting gro file (1yrf_ions.gro) will be used for the simulation.

Simulation
The second phase of this tutorial consists of actually simulating the system we prepared.
The simulation procedure involves multiple steps.
- Energy minimization
- NVT equilibration
- NPT equilibration
- Production run
They are all based on two modules:
- grompp
- mdrun
The general idea is similar to what we saw for the genion module.
We use the grompp module to assemble a tpr input file. The difference is that we run the input file with the mdrun command.
We will need an mdp parameter file for each run to define the simulation settings. The mdp templates are discussed in more detail here.
Energy minimization
This procedure aims to minimize the potential energy of the system by adjusting the atomic coordinates. It will stabilize the overall structure and avoid steric clashes.
Download the mdp parameter file (minim.mdp) from here.
Use the grompp module to generate a tpr file for the energy minimization (em.tpr).
|
|
Now you can run the simulation with the mdrun command.
|
|
Depending on your computational resources this process may take from a few seconds to minutes.
The mdrun module generates four files:
em.gro: containing the final structureem.log: information about the runem.trr: with the trajectory of the systemem.edr: an energy file useful for the analysis of the simulation
We can use this last file to observe how the energy gets minimized along the simulation with the gmx energy module. This module will generate a file in the xvg format that we can use to generate a plot.
|
|
At the prompt, type:
|
|
to select Potential energy.
You can plot the resulting xvg file (potential.xvg) using Grace. Find a more detailed description of the gmx energy module and the xmgrace tool here.
|
|
This should be the result.
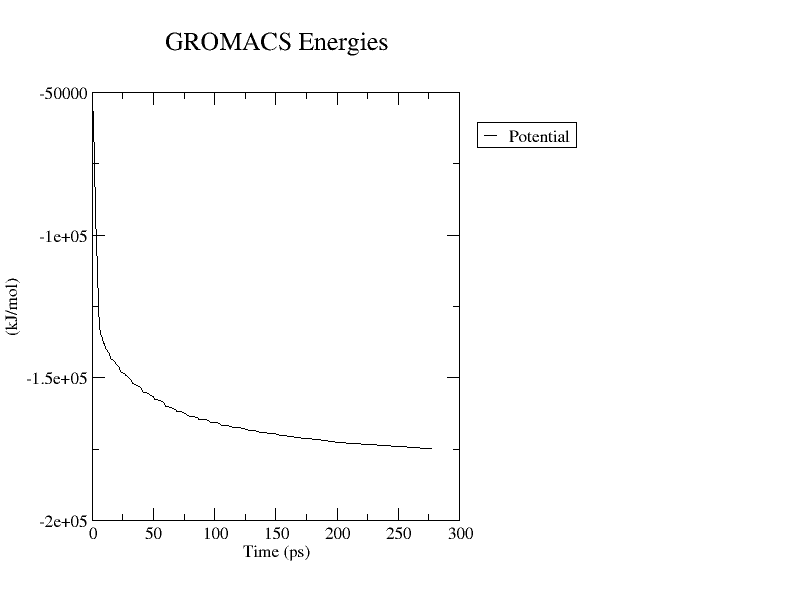
You can also download the initial structure (1yrf_ions.gro) and compare it to the new one (em.gro) by loading both of them in Pymol.
NVT equilibration
The energy minimization ensured that we have a reasonable starting structure, in terms of geometry and solvent orientation. Before proceeding with the actual simulation we need to perform an equilibration procedure. This is needed to equilibrate the ions and the solvent around the protein.
We will divide it in two phases.
The first one will be carried within the NVT ensemble. This means that we are going to implement a thermostat, and we will equilibrate the system at constant temperature.
We are going to run a 100 ps simulation using the V-rescale thermostat.
Find the nvt.mdp file needed to run the grompp module here.
|
|
-r option. The atoms to be restrained are contained in the posre.itp that we initially generated via the gmx pdb2gmx module.
Now you can run the simulation with this command
|
|
Starting from now, the simulations will be more computationally intensive. Depending on the machine you are using this process could take some time.
Once the simulation is over we can use the gmx energy to extract properties from the run.
In NVT equilibration the temperature of the system should reach a plateau at the value we specified in the mdp file. Let’s see how long it takes for the temperature to reach a stable value.
|
|
Plot the xvg file.
|
|

As you can see the temperature is quite stable around the selected value (300K). In general, if the temperature has not yet stabilized, additional time will be required.
To check that the system is properly equilibrated we can check the RMSD value via the gmx rms module.
|
|
This will output an xvg file (nvt_rmsd.xvg) with the backbone to backbone RMSD for the protein. Let’s plot it.
|
|
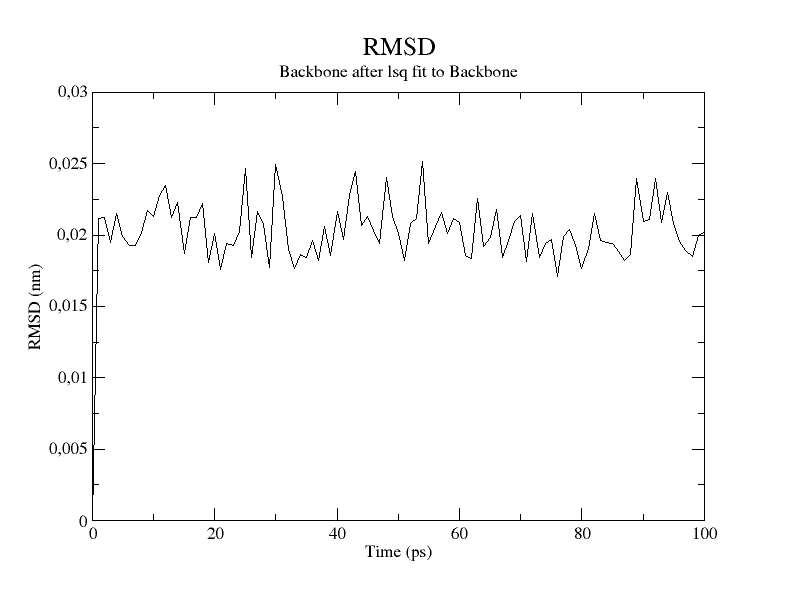
The RMSD quickly converges to a stable value signaling that the system has equilibrated at the desired temperature. We can move to the next phase.
NPT equilibration
The second equilibration will be carried out within the NPT ensemble. In this case, we are going to bring the system at constant pressure through a virtual barostat. We will run a 100 ps simulation using the Parrinello-Rahman barostat.
Download the npt.mdp parameter file here.
Assemble the tpr file with the grompp command, and then launch the simulation with mdrun.
|
|
|
|
We can monitor the progression of the pressure with the gmx energy module as previously shown.
|
|

You can see that the protein widely fluctuates around the pressure value we selected in the mdp file (1 bar). Don’t worry, large pressure fluctuations are completely normal for nanoscale simulations of aqueous solutions.
Once again, we can check the RMSD value to control that the system has properly equilibrated at constant pressure.
|
|

The RMSD values are very stable over time, indicating that the system is well-equilibrated at the desired pressure.
Production run
Now that our system is finally equilibrated at the desired temperature and pressure we are ready for the real simulation. This is the so-called production run.
During this phase, we are going to release the position restraints on the heavy atoms of the protein. We will observe how the system evolves over time, and depending on our goal we will collect relevant data.
The production run will still be carried out within the NPT ensemble using the V-rescale thermostat and the Parrinello-Rahman barostat.
For the purpose of demonstration, we are just going to perform a 1 ns simulation. Keep in mind that in a real experiment you will need to run hundreds of ns to extract useful information from your system.
Find the md.mdp file here.
|
|
|
|
The production run will be a little bit longer than the previous one. If you want to run it in background use:
|
|
This will append the output in a temporary file named nohup.out
You can check the progress of the simulation with the following command:
|
|
When the simulation is over you will still find the relevant informations in the usual files (md.edr, md.log, md.xtc)
Congratulations! you have now successfully conducted your MD simulations. This ends the tutorial.
However, GROMACS comes with a lot of tools to analyze the trajectory of a protein. We already saw some practical examples of the gmx energy and the gmx rms modules. Feel free to use these commands to analyze the results of this run.
Yuo shouldn’t stop here. Analyzing your system is like putting together a puzzle, the more pieces you have the clearer the picture gets.
For this reason, you can find other articles dedicated to a series of analysis you may be interested in performing for your research:
- Analyze the hydrogen bonds with the gmx hbond module
- Compute the RMSF with the gmx rmsf module
- Estimate the Solvent Accessible Surface Area (SASA) via the gmx sasa module
- Cluster your trajectory to find the most representative structure of the simulation
Pro Tip
Here you can find a list of the commands we used to carry out the simulation. It is always useful to have them in a file so that you can simply copy and launch them from the terminal.
Just remember to change the name of the initial pdb file.
In the end, you can find links to the .mdp file templates.
System preparation
|
|
Simulation
|
|