Align structures in PyMOL
As a beginner in computational chemistry, mastering the art of aligning molecular structures is crucial for comparative protein analysis. There are several ways you can use to align two molecular structures, here I will show you how to do it using the PyMOL command line.
Indeed, PyMOL offers powerful alignment capabilities that allow you to compare and contrast different structures, identify structural similarities and differences, and gain insights into protein function.
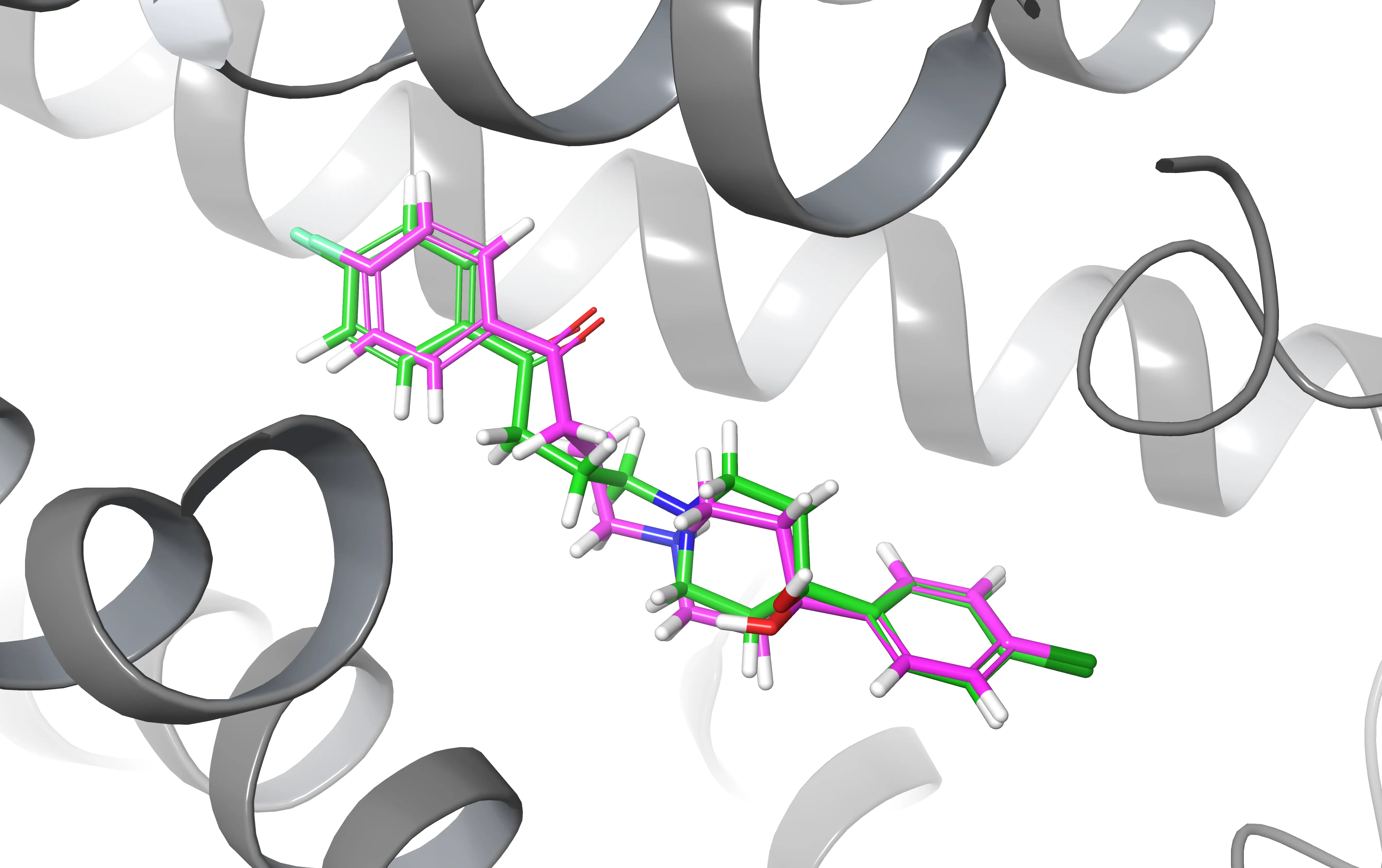
Example Aligning structures can be useful when you want to compare two ligands after a docking experiment
The PyMOL align command
The way this command work is quite intuitive.
Let’s say that you have two structures (object_1, object_2) and you want to align them. You only need to type the following in the PyMOL console.
|
|
Note that the first one will align with the second one. Therefore the first selected object will be the “mobile” one while the second will be the reference.
|
|
PyMOL uses a two-step approach for aligning structures: first, it performs a sequence alignment, and then it minimizes the Root Mean Square Deviation (RMSD) between the aligned residues. As a result, you will get the structures aligned in the display and something like this will be printed to the console.
|
|
Examples of alignments using PyMOL
By utilizing PyMOL’s alignment capabilities, you can perform various alignments to gain insights into the structural characteristics of biomolecules and their interactions. Now let’s look at some use cases you may encounter during your research. Most of them require that you are more or less familiar with the Pymol selection tool so I suggest you read this article if you want to get an idea of how it works.
Align two proteins in pymol
Let’s say you have two protein structures in the PDB format, protein_A.pdb and protein_B.pdb, and you want to align them to analyze their structural similarities and differences. You can follow these two steps:
- Load the protein structures into PyMOL: You can use the
loadcommand in PyMOL to load the protein structures into separate objects. For example:
|
|
This will load the protein structures from the PDB files protein_A.pdb and protein_B.pdb into two separate objects named mobile and reference in the PyMOL GUI.
|
|
- Proceed to align them as previously shown using the
aligncommand:
|
|
You can also use the fetch command to directly retrieve the pdb file from the Protein Data Bank.
|
|

Align specific chains or groups of residues
Sometimes you may notice that the standard alignment is not great. If that is the case, you can improve by selecting a subset of residues and atoms.
Let’s take as an example a situation where you have two proteins and you want to align one protein to a specific chain or certain residues of the other protein. You can use PyMOL’s align command along with the sele command to specify the chains or residues of interest. Here’s an example:
- Load the two proteins
|
|
- Select the chain or residues of interest: You can use the
selecommand to specify the chain or residues of interest that you want to align.
|
|
This will select chain A from object1 as the chain of interest, or residues 100 to 150 from object2, respectively.
- Align the selected chain or residues: You can use the
aligncommand to align the selected chain or residues to the other protein structure.
|
|
Align based on the protein backbone
Similarly, we can align two proteins based on their backbones by first extracting the alpha carbons of the protein with the sele command.
|
|
Align the two objects containing the alpha carbons:
|
|
Align two ligands
Let’s say that you run a MD simulation using GROMACS or any other software and you want to observe how the position of the ligand changes along the course of the simulation.
Something you could do is to load both the initial and final frames (you can get them via gmx trjconv) of the simulation and align two ligands based on their names and residue codes (resn) using the sele command in PyMOL.
|
|
Align the two objects containing the ligands:
|
|
This will align the two ligands based on their residue name “LIG”, minimizing the RMSD between the ligand atoms. After aligning the ligands, you can visually compare the superimposed structures in PyMOL to analyze whether something changed.
Align multiple pdb files
PyMOL also allows you to align multiple structures. To do that, you have to load all the structures and then perform consecutive alignments. A little cumbersome but it gets the job done.
|
|
In this example, we have loaded three PDB files (protein1.pdb, protein2.pdb, and protein3.pdb) into three separate objects (reference, object2, and object3).
We then used the align command to align object2 and object3 to the same reference.